Oyster River Protocol For Transcriptome Assembly¶
The OR Protocol for transcriptome assembly is an actively developed, evidenced based method for optimizing transcriptome assembly. The preprint corresponding to this protocol is here: http://biorxiv.org/content/early/2015/12/30/035642
Contact Information¶
- Gitter (preferred)
- Email (good): Matthew.MacManes@unh.edu
- Twitter (good): @MacManes
- Phone (discouraged): 603-862-4052
- Office (I’m hiding under my desk): 189 Rudman Hall

Some method you’d like me to benchmark? File an issue
How to set up AWS machine for assembly¶
0. Archive Reads.¶
It is likely a good idea to compress your raw reads and save them elsewhere - like another computer. Computers fail, drives corrupt. Better to NOT lose your data in the process.
1. Initial Quality Check¶
SolexaQA++ analysis file_1.fastq file_2.fastq
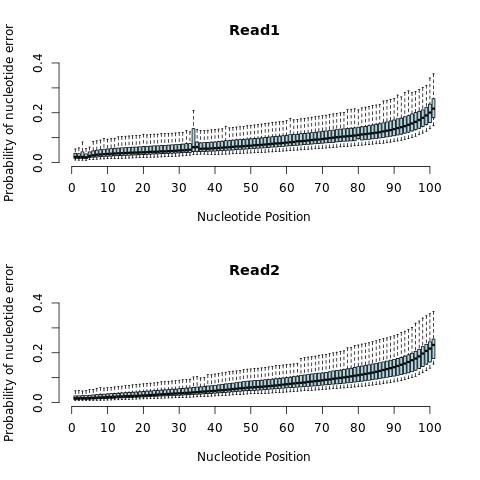
Plot Results using R
R #this opens R on your AWS machine
qual1 <- read.delim("file_1.fastq.quality")
qual2 <- read.delim("file_2.fastq.quality")
jpeg('qualplot.jpg')
par(mfrow=c(2,1))
boxplot(t(qual1), col='light blue', ylim=c(0,.4), frame.plot=F, outline=F, xaxt = "n", ylab='Probability of nucleotide error', xlab='Nucleotide Position', main='Read1')
axis(1, at=c(0,10,20,30,40,50,60,70,80,90,100), labels=c(0,10,20,30,40,50,60,70,80,90,100))
boxplot(t(qual2), col='light blue', ylim=c(0,.4), frame.plot=F, outline=F, xaxt = "n", ylab='Probability of nucleotide error', xlab='Nucleotide Position', main='Read2')
axis(1, at=c(0,10,20,30,40,50,60,70,80,90,100), labels=c(0,10,20,30,40,50,60,70,80,90,100))
dev.off()
quit()
n
2. Error Correct¶
Use RCorrector if you have more than 20 million paired-end reads
run_rcorrector.pl -k 31 -t 30 \
-1 file_1.fastq \
-2 file_2.fastq
Use bfc if you have less than 20 million paired-end reads. If you are using Illumina fastQ format 1.8 or later, read this before attempting BFC correction
seqtk mergepe file_1.fastq file_2.fastq > inter.fq
bfc -s 50m -k31 -t 16 inter.fq > bfc.corr.fq
split-paired-reads.py bfc.corr.fq
mv bfc.corr.fq.1 bfc.corr.1.fq
mv bfc.corr.fq.2 bfc.corr.2.fq
3. Aggressive adapter & gentle quality trimming.¶
One should aggressively hunt down adapter seqeunces and get rid of them. In contrast, gently trim low quality nucleotides. Any more will cause a significant decrease on asembly completeness, as per http://journal.frontiersin.org/article/10.3389/fgene.2014.00013/. I typically do both these steps from within Trinity (using Trimmomatic), but one could do trimming as an independent process if desired.
skewer -l 25 -m pe -o skewer --mean-quality 2 --end-quality 2 -t 30 \
-x /home/ubuntu/share/TruSeq3-PE.fa \
file_1.cor.fastq file_2.cor.fastq
4. Assemble¶
Assemble your reads using Trinity and BinPacker. If you have stranded data, make sure to iclude the --SS_lib_type RF tag, assuming that is the right orientation (If you’re using the standard TruSeq kit, it probably is). Also, you may need to adjust the --CPU and --max_memory settings. Change the name of the input reads to match your read names.
Trinity --seqType fq --max_memory 10G --CPU 16 --output Rcorr_trinity --full_cleanup \
--left skewer-trimmed-pair1.fastq \
--right skewer-trimmed-pair2.fastq
spades.py -o Rcorr_spades --rna \
--only-assembler --threads 16 --memory 20 \
-1 skewer-trimmed-pair1.fastq \
-2 skewer-trimmed-pair2.fastq
5. TransFuse Merge Assemblies¶
Each Assembler will reconstruct a slightly different set of _true_ transcript. TransFuse will take them both and merge them together
transfuse -t 16 -i 0.98 -o transfuse.fasta \
-l skewer-trimmed-pair1.fastq \
-r skewer-trimmed-pair2.fastq \
-a Rcorr_spades/transcripts.fasta,Rcorr_trinity.Trinity.fasta
6. Quality Check¶
If you have followed the ORP AWS setup protocol, you will have the BUSCO Metazoa and Vertebrata datasets. If you need something else, you can download from here: http://busco.ezlab.org/. You should check your assembly using BUSCO. For most transcriptomes, something like 60-90% complete BUSCOs should be accepted. This might be less (even though your transcriptome is complete) if you are assembling a marine invert or some other ‘weird’ organism.
BUSCO.py -m tran --cpu 16 -l ~/busco/eukaryota_odb9 \
-o assemb_name -i transfuse.fasta
You should evaluate your assembly with Transrate, in addition to BUSCO. A Transrate score > .22 is generally thought to be acceptable, though higher scores are usually achievable. There is a good*fasta assembly in the output directory which you may want to use as the final assembly, for further filtering [e.g., TPM], or for something else.
transrate -o assemb_name -t 16 \
-a transfuse.fasta \
--left skewer-trimmed-pair1.fastq \
--right skewer-trimmed-pair2.fastq
7. Filter¶
Filtering is the process through which you aim to maximize the Transrate score, which assays structural integrity, while preserving the BUSCO score, which assays genic completeness. At some level this is a trade off. Some people may require a structually accurate assembly and not care so much abot completeness. Others, dare I say most, are interested in completeness - reconstructing everything possible - and care less about structure.
In general, for low coverage datasets (less than 20 million reads), filtering based on expression, using TMP=1 as a threshold performs well, with Transrate filtering often being too aggressive. With higher coverage data (more than 60 million reads) Transrate filtering may be worthwhile, as may expression filtering using a threshold of TMP=0.5. Again, these are general recommendations, you’re dataset may perform differently.
To do the filtering, run BUSCO on the good*fasta file which is a product of Transrate. This assembly may be very good (or maybe not). I typically use this one if the number of BUSCOs does not decrease by more than a few percent, relative to the raw assembly output from Trinity. Use the BUSCO code from above, changing the name of the input and output. In addition to Transrate filtering (of as an alternative), it is often good to filter by gene expression. I typically filter out contigs whose expression is less than TMP=1 or TMP=0.5.
Estimate expression with Kallisto
kallisto index -i kallisto.idx transfuse.fasta
kallisto quant -t 32 -i kallisto.idx -o kallisto_orig skewer-trimmed-pair1.fastq skewer-trimmed-pair2.fastq
Estimate expression with Salmon
salmon index -t transfuse.fasta -i salmon.idx --type quasi -k 31
salmon quant -p 32 -i salmon.idx --seqBias --gcBias -l a -1 skewer-trimmed-pair1.fastq -2 skewer-trimmed-pair2.fastq -o salmon_orig
Pull down transcripts whose TPM > 1.
awk '1>$5{next}1' kallisto_orig/abundance.tsv | awk '{print $1}' > kallist
awk '1>$4{next}1' salmon_orig/quant.sf | sed '1,10d' | awk '{print $1}' > salist
cat kallist salist | sort -u > uniq_list
python ~/share/filter.py transfuse.fasta uniq_list > Highexp.fasta
8. Annotate¶
I have taken a liking to using dammit! (http://dammit.readthedocs.org/en/latest/).
mkdir ~/dammit/ && cd ~/dammit
dammit databases --install --database-dir ~/dammit --full --busco-group metazoa
dammit annotate Highexp.fasta --busco-group metazoa --n_threads 36 --database-dir ~/dammit/ --full
9. Report¶
Verify the quality of your assembly using content based metrics. Report Transrate score, BUSCO statistics, number of unique transcripts, etc. Do not report meaningless statistics such as N50